|
|
КНИГИ ПО ХИМИИ: |
|
   |
ЧАСТЬ ВТОРАЯСИНТЕЗ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ Галоидопроизводные углеводородов и других органических соединений широко применяются в качестве исходных веществ в ряде синтезов. Относительно большая подвижность атома галоида делает возможным его замещение на различные радикалы - на гидроксил, аминогруппу, цианогруппу, карбоксил и пр. Из разнообразных методов получения галоидопроизводных наибольшее значение имеют:
1. Реакции замещения гидроксила на галоид при нагревании с концентрированными галоидоводородными кислотами или с галоидопроизводными соединений фосфора.
2. Реакции прямого замещения водорода на галоид.
3. Присоединение галоида или галоидоводорода к двойной или тройной связи.
Замещение спиртового гидроксила на галоид Образование бромистого этила происходит в результате следующих реакций:
KBr + H2SO4 а HBr + KHSO4
C2H5-OH + HBr а C2H5Br + H2O
Приведенные реакции, вообще говоря, являются обратимыми, но так как конечный продукт - бромистый этил - все время удаляется из реакционной среды, реакция протекает почти до конца в направлении, указанном стрелками. Для более полного использования бромистоводородной кислоты спирт берут в некотором избытке. К реакционной смеси добавляют небольшое количество воды с целью воспрепятствовать вспениванию реакционной массы, уменьшить образование побочного продукта - этилового эфира - и устранить потери бромистоводородной кислоты за счет улетучивания.
Реактивы:
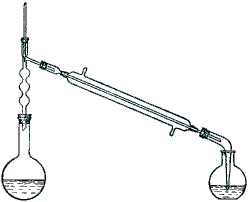
В круглодонную колбу емкостью 300 мл вливают спирт, добавляют 35 мл воды и при постоянном помешивании и охлаждении постепенно приливают 75 мл концентрированной серной кислоты. Смесь охлаждают до комнатной температуры и при перемешивании прибавляют тонко растертый бромистый калий. Колбу соединяют (рис. 28) с дефлегматором и длинным, хорошо действующим холодильником, к которому присоединяют изогнутую насадку. Так как бромистый этил чрезвычайно летуч, то для уменьшения потерь за счет испарения погон собирают в ледяную воду. Для этого в приемник наливают немного воды, бросают несколько кусочков льда и погружают в нее конец насадки.
Реакционную смесь нагревают на песочной или воздушной бане до тех пор, пока в приемник не перестанут переходить маслянистые капли, опускающиеся на дно. Если реакционная смесь в колбе начинает слишком пениться, то на короткое время прерывают нагревание.
По окончании реакции отделяют при помощи делительной воронки бромистый этил от воды, переливают его в небольшую колбу и для освобождения от примесей эфира прибавляют по каплям (при охлаждении в смеси льда и соли и при встряхивании) концентрированную серную кислоту до тех пор, пока она не соберется в виде отдельного слоя под бромистым этилом. Весь эфир растворяется при этом в кислоте*1.
Бромистый этил отделяют при помощи делительной воронки от серной кислоты и, не промывая его водой, перегоняют из перегонной колбы емкостью 100 мл, собирая в приемник, помещенный в охлаждающую смесь.
Выход около 45 г.
Темп. кип. 38,4; уд. вес
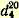 1,4555; показатель преломления 1,4555; показатель преломления 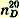 1,4239. 1,4239. Препарат может быть использован для синтеза этилбензола или для синтеза этилмалоновой кислоты. Так как при этих реакциях примесь небольших количеств эфира не имеет значения, то обработку бромистого этила серной кислотой можно не производить, а высушить его (после отделения воды) прокаленным хлористым кальцием и перегнать.
Иодистый этил нельзя получать таким же путем, как бромистый этил, т. е. действием на спирт смеси иодистого калия с серной кислотой, так как образующаяся иодистоводородная кислота восстанавливает серную кислоту, приводя к образованию сернистой кислоты и свободного иода.
Для получения иодистого этила используют реакцию спиртов с трехиодистым фосфором:
3C2H5-OH + PI3 а 3C2H5I + H3PO3
Треххлористый фосфор получают во время самой реакции согласно уравнению:
2P + 3I2 а 2PI3
Так как иод является самым дорогим из реагентов, то для более полного его использования фосфор и спирт берут в небольшом избытке по сравнению с теоретическими количествами.
Реактивы:
В круглодонную колбу емкостью 100-150 мл помещают фосфор и спирт и постепенно при помешивании прибавляют мелко растертый иод. Затем колбу соединяют с обратным холодильником и оставляют стоять 2 часа при частом встряхивании, после чего нагревают в течение 2 час. на водяной бане. Смеси дают охладиться, заменяют обратный холодильник нисходящим и отгоняют иодистый этил на кипящей водяной бане. Под конец отнимают водяную баню и отгоняют остаток иодистого этила, осторожно нагревая колбу на сетке.
В погоне, кроме иодистого этила, содержится некоторое количество спирта и немного иода, окрашивающего дистиллят в коричневый цвет. Для удаления спирта отогнанную жидкость дважды энергично встряхивают в делительной воронке с водой; для удаления иода к воде прибавляют несколько капель раствора бисульфита. Иодистый этил отделяют от воды, сушат прокаленным хлористым кальцием и перегоняют из маленькой перегонной колбы, нагревая ее большим пламенем.
Выход около 30 г.
Иодистый этил - тяжелая бесцветная жидкость со сладковатым запахом; темп. кип. 72,3° уд. вес
1,9371; показатель преломления 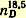 1,5133. 1,5133. Иодистый этил при стоянии на свету темнеет; поэтому его хранят в плотно закупоренной склянке темного стекла над небольшим количеством ртути. Препарат может быть использован для получения фенетола, этилбензола, и этилмалоновой кислоты.
Реакция образования бромистого бутила при действии серной кислоты и бромистого калия на н-бутиловый спирт аналогична описанной выше реакции образования бромистого этила. В данном случае берут некоторый избыток бромистого калия для более полного использования бутилового спирта. К реакционной смеси прибавляют немного воды с целью ослабить течение побочных реакций, в частности образования бутилена, что легко происходит в присутствии концентрированной серной кислоты:
CH3-CH2-CH2-CH2OH а CH3-CH2-CH=CH2 +H2O
Реактивы:н-Бутиловый спирт.....46 мл или 37 г (0,5 моля) Бромистый калий.......75 г (0,63 моля) Серная кислота; бисульфат натрия; бикарбонат натрия; хлористый кальций
В круглодонную колбу емкостью 0,5 л вливают 70 мл воды и прибавляют тонко растертый бромистый калий и бутиловый спирт. Присоединяют обратный холодильник, вставляют в его внутреннюю трубку воронку и через нее небольшими порциями (приблизительно по 5-7 мл) приливают 50 мл концентрированной серной кислоты, каждый раз хорошо перемешивая смесь покачиванием колбы (зажим, удерживающий холодильник, должен быть зажат неплотно).
В колбу бросают несколько кусочков пористой глиняной тарелки и нагревают до кипения на сетке на небольшом пламени в течение 2 час. Затем заменяют обратный холодильник нисходящим и, усилив нагревание, быстро отгоняют бромистый бутил. Сырой продукт содержит примесь воды, бутилового эфира, бутилового спирта, немного бутилена и следы брома. Его промывают в делительной воронке водой, содержащей немного бисульфата; отделив водный слой, переливают бромистый бутил в сухую делительную воронку и для удаления эфира промывают равным объемом холодной концентрированной серной кислоты. Кислоту отделяют, промывая бромистый бутил последовательно чистой водой, разбавленным раствором бикарбоната натрия и снова водой и сушат хлористым кальцием. Полученный продукт перегоняют, собирая фракцию, кипящую в пределах 98-103°.
Выход 50-55 г.
Темп. кип. чистого препарата 101,6° уд. вес 1,299
Препарат может быть использован для синтеза н-октана.
При реакциях прямого замещения водорода на галоид обычно получается смесь различных продуктов. С одной стороны, замещению могут подвергаться атомы водорода, находящиеся у различных атомов углерода, что приводит к образованию смеси изомеров, часто весьма сложной. С другой стороны, замещаться может не один, а последовательно несколько атомов водорода, что приводит к образованию смеси моно-, ди- и тригалоидопроизводных. Поэтому для синтеза галоидопроизводных жирного ряда реакция прямого замещения может быть использована лишь в тех случаях, когда в молекуле исходного продукта один из атомов водорода способен замещаться на атом галоида значительно легче по сравнению с другими. Так, в карбоновых кислотах особенно легко замещается атом водорода при a-атоме углерода; другие изомеры при этом не получаются. Ниже в качестве примера реакции замещения атома водорода при a-атоме углерода кислоты описан синтез монохлоруксусной кислоты.
Более широко используется реакция прямого замещения для синтеза галоидопроизводных ароматического ряда, где удается, соблюдая соответствующие условия, достигнуть замещения атома водорода, находящегося у того или иного определенного атома углерода, и получить достаточно однородный продукт реакции.
Реакция замещения водорода на галоид сильно ускоряется при нагревании, под действием света (особенно ультрафиолетовых лучей) и в присутствии катализаторов; в качестве последних применяют иод, железо, хлористый или бромистый алюминий и др. Прямое замещение водорода на иод может быть осуществлено в том случае, если приняты меры к удалению образующейся при реакции иодистоводородной кислоты так как последняя способна восстанавливать получаемое галоидопроизводное.
Образование монохлоруксусной кислоты, протекающее согласно уравнению:
CH3-COOH +Cl2 а CH2Cl-COOH +HCl
значительно облегчается в присутствии пятихлористого фосфора. Последний образуется в процессе самой реакции при взаимодействии красного фосфора с хлором. Наряду с монохлоруксусной кислотой получается некоторое количество ди- и трихлоруксусной кислоты, от которых основной продукт реакции освобождается перегонкой или кристаллизацией.
Реактивы:
При работе с монохлоруксусной кислотой следует соблюдать осторожность, так как она сильно разъедает кожу.
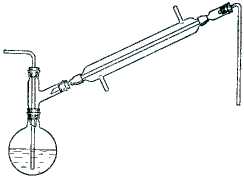
В круглодонную короткогорлую колбу емкостью 100-150 мл помещают смесь ледяной уксусной кислоты и 5 г красного фосфора. При помощи двугорлой насадки колбу соединяют с обратным холодильником. В прямой отросток насадки вставляют стеклянную трубку для пропускания хлора (рис. 29). Прибор устанавливают на хорошо освещенном месте, лучше всего на прямом солнечном свету, так как скорость хлорирования сильно зависит от освещения. Колбу нагревают на кипящей водяной бане и пропускают сильную струю хлора из баллона (нужно по возможности избегать резиновых соединений, так как резина быстро разрушается хлором). Выделяющийся при реакции хлористый водород и непрореагировавший хлор или поглощают раствором щелочи, или при помощи стеклянной трубки отводят в хорошо действующую тягу. Пропускание хлора ведут до тех пор, пока охлажденная проба (взятая в пробирку) не будет застывать при потирании стеклянной палочкой стенок пробирки. В зависимости от интенсивности освещения реакция заканчивается через 6-12 час.
Продукт реакции перегоняют под тягой из перегонной колбы с воздушным холодильником. Сначала перегоняется хлористый и неизмененная уксусная кислота, а в пределах 150-200° переходит монохлоруксусная кислота. Эту фракцию собирают отдельно и охлаждают ее ледяной водой. При помешивании стеклянной палочкой монохлоруксусная кислота закристаллизовывается; образовавшиеся кристаллы быстро отфильтровывают на воронке Бюхнера и на фильтре же отжимают пестиком или шпателем. Фильтровать надо быстро, так как кристаллы монохлоруксусной кислоты постепенно расплываются на воздухе. Фильтрат перегоняют еще раз, собирая фракцию, кипящую в пределах 170-200° при охлаждении ее удается получить еще некоторое количество кристаллов монохлоруксусной кислоты. Их присоединяют к первой порции кристаллов и перегоняют еще раз, собирая фракцию, кипящую в пределах 155-190°.
Выход 30-45 г.
Монохлоруксусная кислота кипит при 189° при охлаждении застывает в кристаллы, плавящиеся при 61,5°. Она легко растворима в воде; кристаллы ее расплываются во влажном воздухе.
Монохлоруксусная кислота может быть использована для синтеза гликоколя.
Замещение атома водорода в бензольном ядре атомом брома
C6H6 + Br2 а C6H5Br + HBr
идет относительно легко, особенно в присутствии катализаторов, в качестве которых чаще всего применяют железо или алюминий.
Наряду с монобромбензолом получается небольшое количество о- и п-дибромбензола, от которых (как имеющих более высокую температуру кипения) основной продукт реакции может быть освобожден перегонкой. Для более полного использования брома бензол берут в небольшом избытке.
Реактивы:
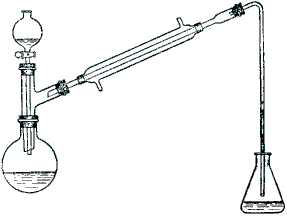
Круглодонную колбу емкостью 150-200 мл при помощи двугорлой насадки соединяют с капиллярной воронкой и обратным холодильником. К холодильнику присоединяют изогнутую трубку, конец которой опускают в колбу с водой, служащей для поглощения образующегося при реакции бромистого водорода (рис. 30). Конец трубки должен находиться над поверхностью воды, в противном случае вода будет втянута в прибор.
В колбу вносят 0,5 г железных опилок, приливают бензол и затем постепенно, при помешивании, прибавляют бром через капельную воронку. Обычно реакция начинается не сразу; поэтому вначале не следует прибавлять слишком много брома. После того как начнется выделение бромистого водорода, приливают остальное количество брома, притом с такой скоростью, чтобы реакция не стала слишком бурной. Для окончания реакции колбу непродолжительное время нагревают на водяной бане.
Подученный продукт переносят в делительную воронку, промывают водой, разбавленным раствором едкого натра, еще раз водой, переливают в сухую колбу, сушат прокаленным хлористым кальцием, после чего перегоняют из небольшой перегонной колбы, применяя воздушный холодильник. Фракцию, перешедшую в пределах 140-170°, перегоняют вторично, собирая фракцию, кипящую в пределах 154-160°.
Выход около 13 г.
Чистый бромбензол представляет собой тяжелую жидкость, кипящую при 156,2° уд. вес
1,4914; показатель преломления 1,5598. В остатке, не перегнавшемся при 170°, содержится о- и п-дибромбензол. Для выделения последнего еще горячую жидкость выливают на часовое стекло и после застывания отжимают кристаллы между листами фильтровальной бумаги (для отделения жидкого о-бромбензола); перекристаллизацией из небольшого количества кипящего спирта получают чистый п-дибромбензол в виде крупных бесцветных кристаллов, плавящихся при 89°.
Выход около 1 г.
a-Бромнафталин получается в результате бромирования нафталина
C10H8 + Br2 а C10H7Br + HBr
Бромирование можно проводить или в водной среде, или же в органическом растворителе; в последнем случае получаются несколько лучшие выходы. В качестве растворителей применяют четыреххлористый углерод или ледяную уксусную кислоту.
а) Бромирования нафталина в водной среде
Реактивы:
В стакане емкостью 200 мл нагревают тонко растертый нафталин с 40 мл воды до 40-50° и при энергичном перемешивании, следя за тем, чтобы поддерживалась указанная температура, добавляют по каплям бром из капельной воронки; трубка последней должна доходить до дна стакана. После того как весь бром прибавлен, продолжают перемешивание до тех пор, пока не исчезнет окраска брома. Выделившееся тяжелое масло отделяют при помощи делительной воронки, переносят в колбу емкостью 150-200 мл и перегоняют с перегретым до 130° паром, пока на соберется 70-90 мл дистиллята. Колбу при этом целесообразно подогревать на масляной бане, нагретой до 145-150°. С перегретым паром удаляется большая часть не вступившего в реакцию нафталина.
Остаток в колбе высушивают хлористым кальцием и перегоняют в вакууме. Бромнафталин перегоняется в пределах 132-135° при остаточном давлении 12 мм в пределах 145-148° при остаточном давлении 20 мм. Из нижекипящей фракции при охлаждении выделяется нафталин, а из вышекипящей - дибромнафталин. Если жидкость отделить отсасыванием от кристаллов, соединить и перегнать еще раз в вакууме, то можно выделить еще некоторое количество бромнафталина.
Выход 20-21 г.
Темп. кип. 281,1° уд. вес
1,4865; показатель преломления 1,6582. Препарат может быть использован для синтеза a-нафтойной кислоты.
б) Бромирование нафталина в уксусной кислоте
Реактивы:
В перегонную колбу емкостью 50 мл помещают нафталин и 25 мл ледяной уксусной кислоты; колбу закрывают пробкой с маленькой капельной воронкой, конец трубки которой должен быть погружен в жидкость. Для поглощения выделяющегося бромистого водорода к отводной трубке колбы присоединяют изогнутую стеклянную трубку; конец ее опускают в колбу с водой, следя за тем, чтобы он находился над поверхностью воды.
Колбу нагревают на кипящей водяной бане и постепенно, при помешивании, начинают приливать бром через капиллярную воронку. Спустя некоторое время начинается выделение бромистого водорода. Приливание брома ведут с такой скоростью, чтобы бромистый водород не увлекал за собой паров брома. Примерно через 20 мин. приливание брома заканчивают и после этого колбу нагревают на кипящей бане еще 3-4 часа, до почти полного прекращения выделения бромистого водорода.
По окончании реакции колбу соединяют с воздушным холодильником и, заменив капельную воронку термометром, отгоняют избыток уксусной кислоты. Перегонку следует вести под тягой, так как при этом выделяется бромистый водород, в качестве приемника применяют мерный цилиндр. После того как отгонится около 20 мл уксусной кислоты*2, перегонку прерывают и удаляют воздушный холодильник, дальнейшую перегонку ведут без холодильника. В пределах 120-220° отгоняются остаток уксусной кислоты и большая часть не вошедшего в реакцию нафталина. При 220-270° переходит небольшое количество нафталина в смеси с бромнафталином. Когда температура достигнет 270°, меняют приемник, усиливают нагревание и в пределах 270-282° перегоняют бромнафталин.
Выход 12-13 г.
Перегнанный при атмосферном давлении бромнафталин темнеет при стоянии, поэтому лучше еще раз перегнать его в вакууме. В этом случае в качестве приемника следует взять маленькую колбу Кляйзена, из которой, не переливая продукт, возможно вести перегонку в вакууме. Целесообразно также, отогнав большую часть уксусной кислоты при атмосферном давлении, дальнейшую перегонку провести в вакууме, собирая фракцию, переходящую в пределах 140-150° при 20 мм остаточного давления.
В остатке от перегонки содержится 1,4-дибромнафталин. Его можно очистить перекристаллизацией из четыреххлористого углерода. Чистый препарат плавится при 79-80°.
Направление реакции взаимодействия гомологов бензола с галоидами зависят от условий опыта. При умеренной температуре, в отсутствие прямого солнечного света и при наличии соответствующих катализаторов (железа, амальгамированного алюминия) происходит замещение в ядре. При нагревании же и при действии солнечного света замещение идет почти исключительно в боковой цепи.
Образование хлористого бензила идет согласно уравнению:
C6H5-CH3 + Cl2 а C6H5-CH2Cl +HCl
Скорость этой реакции сильно повышается с повышением интенсивности освещения: ускоряющее влияние оказывает также пятихлористый фосфор.
Реактивы:
Прибор собирают по схеме, приведенной при описании получения монохлоруксусной кислоты. Для пропускания хлора берут более широкую трубку и в нее помещают термометр, шарик которого должен находиться ниже уровня жидкости в колбе. Чтобы термометр не выпадал из трубки, ее на высоте 1 см от конца слегка сплющивают. Для этого нагревают трубку до размягчения и осторожно сжимают стекло щипцами.
Толуол, к которому добавляют 2,5 г пятихлористого фосфора, нагревают до кипения на воздушной бане или на сетке и пропускают на солнечном свету сильную струю сухого хлора до тех пор, пока температура реакционной смеси на достигнет 160°. В зависимости от интенсивности освещения реакция заканчивается в течение 1-6 час. По окончании реакции прекращают пропускание хлора и нагревание из той же колбы (под тягой) перегоняют продукт реакции, присоединив к колбе небольшой дефлегматор и длинный воздушный холодильник. Сначала перегоняется на прореагировавший толуол (кипит при 111°); когда температура достигнет 160°, меняют приемник и собирают фракцию, кипящую в пределах 160-190° и содержащую основную массу хлористого бензила. Вышекипящие фракции содержат хлористый бензилиден C6H5CHCl и бензотрихлорид C6H5CCl3. Среднюю фракцию еще раз перегоняют с дефлегматором, собирая продукт, переходящий при 170-180° и представляющий собой почти чистый хлористый бензил.
Выход около 35-40 г.
Более чистый препарат можно получить, перегнав полученный продукт еще раз в вакууме; собирают фракцию, перегоняющуюся при 63-68° при 12 мм остаточного давления.
Чистый хлористый бензил представляет собой бесцветную жидкость; действует раздражающе на слизистые оболочки глаз. Темп. кип. 179,4°; уд. вес
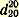 1,1002; показатель преломления 1,1002; показатель преломления 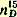 1,5415. 1,5415. Препарат может быть использован для синтеза фенилуксусной кислоты.
Атом галоида в боковой цепи обладает значительно большей подвижностью по сравнению с атомом галоида в ядре. Это различие легко обнаружить следующим опытом: в двух пробирках нагревают по нескольку капель хлористого бензила и бромбензола со спиртовым раствором азотнокислого серебра. Раствор хлористого бензила быстро мутнеет вследствие образования хлористого серебра; бромбензол с азотнокислым серебром не реагирует.
Образование хлористого бензилидена идет согласно уравнению:
C6H5-CH3 + 2Cl2 а C6H5-CHCl2 + 2HCl
Реактивы:
Синтез проводят совершенно так же, как и получение хлористого бензила; хлор пропускают до тех пор, пока температура кипения не достигнет 190°, на что требуется при достаточно хорошем освещении около 6 час. Продукт реакции представляет собой смесь неизмененного толуола, хлористого бензила, хлористого бензилидена и бензотрихлорида. Смесь перегоняют с дефлегматором, и фракцию, перешедшую в пределах 160-225°, перегоняют еще раз, собирая чистый продукт, кипящий при 180-220°.
Выход около 35 г.
Чистый хлористый бензилиден кипит при 205,2°, его уд. вес
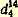 1,2557. 1,2557. Препарат может быть использован для получения бензойного альдегида.
При взаимодействии непредельных углеводородов с бромом получаются с хорошими выходами соответствующие дибромпроизводные (дибромиды).
Непредельные углеводороды, являющиеся исходными веществами для синтеза дибромидов, обычно получают одновременно из соответствующих спиртов. При нагревании последних с водуотнимающими средствами, например с концентрированной серной кислотой или безводным хлористым цинком, происходит отщепление воды и образуется непредельные углеводороды. Еще более гладко отщепление воды от спиртов происходит при пропускании их паров над нагретыми до 360-450° окисью алюминия или глиной (катализаторы).
Образование бромистого этилена из этилового спирта протекает согласно уравнениям:
CH3-CH2-OH а CH2=CH2 + H2O
CH2=CH2 + Br2 а CH2Br-CH2Br
а) Получение этилена путем каталитической дегидратации этилового спирта.
Реактивы:
В качестве катализаторов реакции дегидратации могут быть использованы или окись алюминия, или обычная (необожженная) глина. Их помещают в тугоплавкую стеклянную трубку диаметром 15-18 мм и длиной 60 см, через которую пропускают при нагревании пары спирта. Чтобы катализатор не высыпался из трубки, в нее вставляют на расстоянии 8 см от концов неплотные пробки из асбеста. Окись алюминия насыпают в трубку слоем высотой не более 1/3 диаметра последней. Если в качестве катализатора пользуются глиной, то ее смешивают с небольшим количеством воды, растирают до образования небольших неплотных комочков и подсушивают на умеренно нагретой песочной бане или в сушильном шкафу.
Высушенными кусочками глины наполняют трубку для дегидратации.
Заполненную катализатором трубку вставляют в специальную трубчатую печь с электрическим обогревом. Нагревание печи регулируют при помощи реостата. Для измерения температуры пользуются термопарой с гальванометрами. Очень удобно следить за нагреванием печи по амперметру если заранее известно, какой температуре печи соответствует та или иная сила тока. Этот способ наблюдения за температурой, конечно, недостаточно точен, но вполне пригоден для практических целей.
Нужную для работы печь можно легко изготовить в лаборатории своими силами. Для этого железную трубку длиной 45 см и диаметром около 30 мм обвертывают тонким слоем влажного асбестового картона. Концы трубки обматывают толстым слоем асбеста, на который туго накладывают несколько витков толстой медной проволоки. К медной проволоке прикрепляют концы нагревательной спирали (никелин, нихром и т. п.), равномерными витками которой по влажному асбесту обматывают всю трубку. Вблизи концов трубки витки следует положить несколько чаще, чем в середине трубки. Концы медной проволоки присоединяют к подводящим ток проводам, а всю трубку в целях теплоизоляции обматывают толстым слоем асбестового картона, который закрепляют несколькими витками проволоки. Перед тем как пользоваться трубкой, ей дают хорошо просохнуть. Печь получается более долговечной, если в качестве изолирующего материала вместо асбеста применить слюду. При напряжении в сети в 120 вольт проволока должна иметь сопротивление около 24 ом при длине около 3 м.
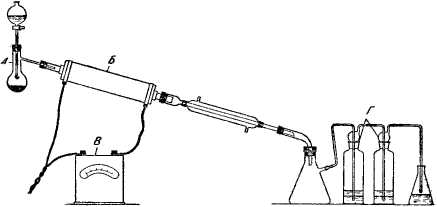
Прибор собирают, как показано на рис. 31. В маленькую перегонную колбу, служащую для испарения спирта, насыпают слой крупного песка и закрывают колбу резиновой пробкой, в которую вставлена капельная воронка; к ножке воронки при помощи резиновой трубки присоединяют стеклянную трубку с оттянутым и загнутым кверху концом. Отводную трубку перегонной колбы укрепляют при помощи резиновой пробки к трубке с катализатором. Трубку с катализатором соединяют с нисходящим холодильником, к которому присоединена колба для отсасывания (или перегонная колба), служащая для сбора образующейся при реакции воды и непрореагировавшего спирта. Тубус колбы для отсасывания соединяют с одной или двумя промывными склянками, содержащими бром и небольшое количество воды (вода должна образовывать над бромом слой в 1 см). Для лучшего охлаждения эти склянки ставят в холодную воду. За промывными склянками ставят колбу с раствором едкого натра для поглощения паров брома (трубка, отводящая пары, не должна быть погружена в щелочь во избежание засасывания ее в склянки с бромом)*3. Перед работой надо проверить весь прибор на плотность соединений.
Включив ток, одновременно начинают нагревать на песочной бане колбу, служащую для испарения спирта. Когда трубка с катализатором нагреется до 370°, в перегонную колбу начинают приливать по каплям спирт из капельной воронки. Капли спирта, падая на нагретый песок, быстро испаряются, и пары спирта поступают в трубку с катализатором.
Газ, образующийся при дегидратации спирта, должен почти полностью поглощаться раствором брома.
Для контроля за течением реакции отводную трубку от прибора (опущенную в колбу с раствором едкого натра) периодически погружают в жидкость; при правильной работе из прибора только изредка должны выходить пузырьки газа. Более частое появление пузырьков указывает на слишком высокую температуру трубки с катализатором.
Когда взятое количество брома полностью вступит в реакцию (что узнается по обесцвечиванию брома), промывные склянки отъединяют от прибора и нагревание прекращают. Содержимое промывных склянок переливают в делительную воронку, отделяют бромистый этилен, промывая его водой, разбавленным раствором щелочи, еще раз водой и тщательно отделяют от последней. Высушив продукт хлористым кальцием, его перегоняют, собирая фракцию, переходящую в пределах 129-132°.
Выход около 30 г. Темп. кип. чистого бромистого этилена 131° уд. вес
2,1816; показатель преломления 1,5379. Препарат может быть использован для получения этилендиамина.
Аналогично могут быть получены бромистый пропилен и бромистый бутилен (или изобутилен). Темп. кип. бромистого пропилена 142°, бромистого бутилена 166°, бромистого изобутилена 148-149°.
Большой интерес представляет получение этилена из бромистого этилена по А. П. Сабанееву действием цинковой пыли спирта. Интересная реакция действия цинковой пыли и спирта на дибромиды различного строения тщательно изучена Н. Я. Демьяновым в применении к дибромидам с открытой цепью, а А. Е. Успенским - в применении к дибромидам циклического строения полиметиленового ряда. Важное значение для получения непредельных углеводородов из гидроксильных производных имеет метод дегидратации последних по Н. Д. Зелинскому действием безводной щавелевой кислоты.
б) Получение этилена взаимодействием спирта с серной кислотой.
При взаимодействии спирта с концентрированной серной кислотой сначала образуется этилсерная кислота:
CH3-CH2-OH + HO-SO3H а CH3-CH2-O-SO3H + H2O
При температуре выше 170° этилсерная кислота разлагается с образованием этилена и серной кислоты:
CH3-CH2-O-SO3H а CH2=CH2 + H2SO4
Одновременно протекает побочная реакция - окисление спирта серной кислотой; в результате ее образуется ряд продуктов - окись углерода, сернистый газ и др.
Реактивы: 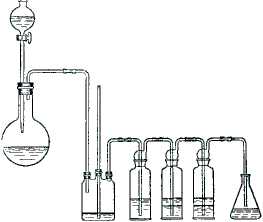 В круглодонную колбу емкостью 300-500 мл вливают 15 мл спирта и затем осторожно, при помешивании, прибавляют 45 мл серной кислоты. Далее для уменьшения вспенивания реакционной смеси насыпают в колбу около 20 г крупного сухого песку, отсеянного от мелких частиц. Полезно также прибавить безводного сернокислого алюминия, действующего каталитически. Колбу закрывают пробкой с двумя отверстиями, в одно из которых вставлена газоотводная трубка, а в другое - капельная воронка с оттянутым в капилляр концом (рис. 32).
Колбу нагревают на сетке или, лучше, на воздушной бане. Как только начнется выделение этилена, через воронку начинают медленно прибавлять смесь из 30 мл спирта и 45 мл серной кислоты. При этом нужно следить, чтобы трубка капельной воронки была целиком заполнена этой смесью.
Выделяющийся этилен пропускают через предохранительную трехгорлую склянку, на дно которой налито немного воды, затем через промывную склянку с 10%-ным раствором едкого натра для поглощения сернистого газа. Этилен, очищенный от сернистого газа, пропускают через две промывные склянки с бромом, как указано выше (получение этилена путем каталитической дегидратации спирта); отводящую трубку от прибора опускают в колбу, содержащую раствор едкого натра.
Нужно обратить внимание на тщательную сборку прибора и проверить его герметичность. Во время работы уровень воды в трубке предохранительной склянки должен быть на 15-20 см выше уровня воды в самой склянке. Если это не наблюдается, нужно проверить, насколько плотно закрыта пробкой реакционная колба, и надежность других соединений.
Пропускание этилена ведут до тех пор, пока бром не вступит полностью в реакцию. Сырой бромистый этилен обрабатывают как указывалось выше, при описании каталитического получения этилена. Выход тот же или лишь немного меньше, чем при каталитическом разложении спирта.
в) Получение этилена взаимодействием спирта с фосфорной кислотой
Для отщепления воды от спиртов может быть использована концентрированная фосфорная кислота. Реакция при этом идет более гладко и этилен получается значительно более чистым, так как отсутствуют окислительные процессы, вызываемые серной кислотой. Продажная фосфорная кислота должна быть предварительно обезвожена нагреванием в фарфоровой чашке до 220° при постоянном помешивании.
Реактивы: В колбу емкостью 250 мл помещают 80 мл обезвоженной фосфорной кислоты и закрывают колбу резиновой пробкой с тремя отверстиями, в которые вставлены газоотводная трубка, капельная воронка с оттянутым концом и термометр. Шарик термометра должен быть погружен в жидкость. Колбу нагревают на сетке и, когда фосфорная кислота нагреется до 210°, начинают понемногу приливать спирт, наблюдая за тем, чтобы температура не превышала 220°.
Выделяющийся этилен через предохранительную склянку пропускают прямо в промывные склянки с бромом; промывная склянка с раствором щелочи в этом случае не нужна. В остальном работу проводят, как указывалось выше, при описании получения этилена путем каталитической дегидратации или действием серной кислоты на спирт.
Хлорангидриды карбоновых кислот легко получаются при взаимодействии кислот с пятихлористым фосфором:
R-C-OH + PCl5 а R-C-Cl + POCl3 + HCl
Атом хлора в хлорангидридах кислот отличается большой подвижностью; поэтому хлорангидриды находят широкое применение при различных синтезах. В частности, хлорангидриды ароматических кислот часто используют для выделения и идентификации спиртов, а также первичных и вторичных аминов, с которыми они образуют ацильные производные, в большинстве случаев мало растворимые и хорошо кристаллизующиеся.
Образование хлористого бензоила протекает согласно уравнению:
C6H5-C-OH + PCl5 а C6H5-C-Cl + POCl3 + HCl
Реактивы: Работу проводят в хорошо действующем вытяжном шкафу.
В круглодонной колбе емкостью 100 мл смешивают бензойную кислоту с порошкообразным пятихлористым фосфором и закрывают резиновой пробкой с вставленной в нее стеклянной трубкой. Колбу встряхивают до тех пор, пока реакционная смесь не станет вполне жидкой. Образовавшуюся смесь хлористого бензоила и хлорокиси фосфора (с примесью небольшого количества пятихлористого фосфора) разделяют перегонкой, применяя небольшой дефлегматор и воздушный холодильник (под тягой). Сначала отгоняется хлорокись фосфора, кипящая при 110°, затем температура повышается и при 199° перегоняется хлористый бензоил.
Выход около 10-11 г.
Температура кипения хлористого бензоила 197,2°.
Хлористый бензоил необходимо сохранять в хорошо закупоренной склянке, так как он разлагается под действием влаги воздуха.
ФенилбензоатC6H5-C-Cl + HO-C6H5 + NaOH а C6H5-C-O-C6H5 + NaCl + H2O
В пробирке растворяют около 0,5 г фенола в 5 мл воды, прибавляют около 0,5 мл хлористого бензоила и 10%-ый раствор едкого натра до щелочной реакции. Смесь слабо подогревают при постоянном перемешивании. Выделившееся масло при охлаждении и потирании стеклянной палочкой закристаллизовывается. Кристаллы отсасывают на маленьком фильтре и перекристаллизовывают из спирта.
Темп. пл. 68-69°.
Гиппуровая кислота Гиппуровая кислота, или бензоилгликоколь, легко образуется при действии хлористого бензоила на гликоколь в присутствии щелочи.
C6H5-C-Cl + H2N-CH2-COOH + 2NaOH а C6H5-C-NH-CH2-COONa + NaCl + 2H2O
В пробирке растворяют в 5 мл воды 1 г гликоколя и прибавляют 1,5 мл хлористого бензоила и 10%-ный раствор едкого натра до щелочной реакции. Смесь энергично перемешивают, добавляя периодически щелочь для поддержания щелочной реакции. Полное исчезновение запаха хлористого бензоила является признаком конца реакции. Содержимое пробирки переливают в небольшую коническую колбочку, охлаждают льдом и подкисляют концентрированной соляной кислотой. Выпавшую гиппуровую кислоту, содержащую примесь бензойной кислоты, отсасывают, тщательно отжимают и для удаления бензойной кислоты промывают несколько раз малыми порциями эфира. Затем перекристаллизовывают из воды, кристаллы отсасывают, отжимают и сушат.
Темп. пл. 187,5°.
Образование хлористого п-нитробензоила протекает согласно уравнению:
O2N-C6H4-C-OH + PCl5 а O2N-C6H4-C-Cl + POCl3 + HCl
Реактивы: Работу проводят в хорошо действующем вытяжном шкафу.
В колбе Кляйзена емкостью 100 мл смешивают п-нитробензойную кислоту с пятихлористым фосфором; оба горла колбы закрывают резиновыми пробками, а к отводной трубке присоединяют изогнутую стеклянную трубку, конец которой опускают в колбу с водой для поглощения выделяющегося хлористого водорода. Надо следить, чтобы конец трубки находился над поверхностью воды, - в противном случае вода будет втянута в реакционную колбу.
Колбу помещают на водяную баню и нагревают до начала реакции. Примерно через 15-20 мин. реакция заканчивается и содержимое колбы превращается в однородную жидкость. Образовавшуюся смесь хлористого п-нитробензоила и хлорокиси фосфора разделяют перегонкой, применяя воздушный холодильник. Сначала отгоняют хлорокись фосфора на масляной бане*4, нагретой до температуры 200-220° (термометр должен быть погружен в масло). Оставшуюся жидкость перегоняют в вакууме; сначала отгоняется небольшое количество хлорокиси фосфора, после чего меняют приемник и продолжают перегонку. При 155° и 20 мм остаточного давления перегоняется хлористый п-нитробензоил, застывающий в желтую кристаллическую массу, плавящуюся при 71°.
Выход около 16 г.
Этил-п-нитробензоатC2H5-OH + Cl-C-C6H4-NO2 а C2H5-O-C-C6H4-NO2 + HCl
В пробирке к 1 мл этилового спирта прибавляют 1 г хлористого п-нитробензоила и нагревают в течение некоторого времени на водяной бане. По охлаждении кристаллическую массу размешивают с разбавленным раствором соды, отсасывают и промывают водой. После перекристаллизации из петролейного эфира или легкого бензина получают чистый п-нитробензоат с темп. пл. 57°.
Аналогичным образом могут быть получены сложные эфиры п-нитробензойной кислоты с другими спиртами. Метиловый эфир плавится при 96°, пропиловый - при 35°.
b-Нафтил-п-нитробензоатC10H7-OH + Cl-C-C6H4-NO2 + NaOH а C10H7 + O-C-C6H4-NO2 + NaCl + H2O
В пробирке к смеси 0,3 г b-нафтола и 0,5 г хлористого п-нитробензоила прибавляют 25 мл 4%-ного раствора едкого натра и 4 мл воды и нагревают при перемешивании. Примерно через 15 мин. выпадают кристаллы п-нитробензоата в виде желтых игл. По охлаждении кристаллы отсасывают, промывая водой и перекристаллизовывают из горячего 95%-ного спирта или, лучше, из ледяной уксусной кислоты. Темп. пл. 166°.
Во многих случаях для получения п-нитробензоатов удобно применять раствор хлористого п-нитробензоила в сухом ацетоне. К этому раствору прибавляют ацилируемое вещество, нагревают некоторое время, а затем прибавляют разбавленный раствор соды до щелочной реакции (по лакмусу). Выпавшие кристаллы отсасывают, промывают водой и перекристаллизовывают из легкого бензина, спирта или другого подходящего растворителя.
Сложный эфир п-нитробензойной кислоты и бензилового спирта плавится при 84°, сложный эфир фенола - при 91°, м-крезола - при 51°, о-крезола - при 90° и п-крезола - при 88°.
*1Этот раствор может быть использован для получения бромистого этила.
*2Отогнанная уксусная кислота содержит небольшие количества бромистого водорода и нафталина и может быть повторно использована при получении бромнафталина.
*3Еще более удобно для поглощения паров брома применить склянку Тищенко с раствором щелочи.
*4Нагревать на голом огне нельзя, так как перегрев вызывает разложение хлористого п-нитробензоила, иногда очень бурное.
|