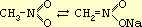|
|
КНИГИ ПО ХИМИИ: |
|
   |
|
Нитросоединения жирного ряда могут быть получены при непосредственном действии азотной кислоты или окислов азота на предельные углеводороды. Прямое нитрование углеводородов жирного ряда и нафтеновых углеводородов изучено М. И. Коноваловым (реакция Коновалова). С. С. Наметкин объяснил механизм этой реакции и широко использовал ее для установления строения терпеновых углеводородов. Однако метод прямого нитрования мало пригоден для препаративных целей, так как он не дает возможности получить достаточно однородный продукт. Лучшие результаты получаются при действии азотистокислых солей на галоидопроизводные углеводородов, например:
C2H5I + AgNO2 а C2H5NO2 + AgI
В качестве побочного продукта при этом образуется некоторое количество сложного эфира азотистой кислоты C2H5-O-NO, который легко отделить от нитросоединения ввиду значительно более низкой температуры его кипения.
Нитросоединения ароматического ряда, напротив, легко получаются при непосредственном нитровании углеводородов и других ароматических соединений - фенолов, кислот и пр. В случае легко нитрующихся соединений реакция идет при применении разбавленной азотной кислоты; трудно вступающие в реакцию вещества нитруют смесью концентрированных азотной и серной кислот. Серная кислота связывает образующуюся при реакции воду и тем самым поддерживает необходимую для реакции концентрацию азотной кислоты. Непосредственное нитрование является единственным практически применяемым методом получения нитросоединений ароматического ряда.
Примером получения нитросоединений жирного ряда действием азотистокислых солей на галоидные производные может служить синтез нитрометана. В качестве исходного вещества берут монохлоруксусную кислоту, в которой хлор обладает значительной подвижностью. При взаимодействии натриевой соли хлоруксусной кислоты с азотнокислым натрием образуется натриевая соль нитроуксусной кислоты:
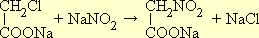 При нагревании нитроуксусная кислота легко декарбоксилируется (т. е. отщепляет CO2), образуя нитрометан:
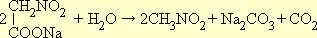
К смеси указанного количества хлоруксусной кислоты и 20 г толченого льда прибавляют при помешивании 14 мл охлажденного во льду 40%-ного раствора едкого натра до щелочной реакции (по фенолфталеину). При нейтрализации температура смеси не должна подниматься выше 20°. Полученный раствор хлоруксуснокислого натрия приливают к раствору азотистокислого натрия в 18 мл воды, находящемся в перегонной колбе емкостью 200 мл. Колбу соединяют с холодильником и закрывают пробкой с термометром, шарик которого должен быть погружен в жидкость.
Смесь медленно нагревают до начала выделения пузырьков углекислого газа, что происходит при температуре около 80°. После этого прекращают нагревание, так как реакция продолжается самостоятельно, без подогрева извне. Если температура смеси начинает понижаться, то вновь осторожно подогревают до 85°. Когда реакция в основном закончится, реакционную смесь осторожно подогревают, доводя температуру ее к концу реакции до 110°. Нитрометан начинает перегоняться при температуре около 90°. При отгонке получают не менее 5 мл нитрометана и около 15 мл воды.
Прекратив перегонку, отделяют при помощи делительной воронки нитрометан, находящийся в нижнем слое, сушат его небольшим количеством хлористого кальция и перегоняют из маленькой перегонной колбы, собирая фракцию, кипящую в пределах 98-102°.
Выход 4-4,5 г.
Темп. кип. чистого нитрометана 101,2°; уд. вес
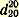 1,1382; показатель преломления 1,1382; показатель преломления 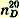 1,3935. 1,3935.
Нитробензол получается при действии на бензол смеси азотной и серной кислот:
C6H6 + HO-NO2 а C6H5-NO2 + H2O
Обязательным условием успешного протекания реакции является хорошее перемешивание реагирующих веществ.
Наряду с нитробензолом образуется небольшое количество динитробензола. Повышение температуры во время нитрования приводит к образованию значительных количеств динитропродукта.
В колбе емкостью 260 мл осторожно, при охлаждении, смешивают азотную кислоту с серной. К охлажденной до комнатной температуры смеси постепенно, небольшими порциями, прибавляют бензол, каждый раз хорошо перемешивая содержимое колбы и наблюдая за тем, чтобы температура смеси не превышала 50-60°. В случае необходимости колбу охлаждают водой. (Для уменьшения потерь бензола за счет испарения к колбе присоединяют воздушный холодильник.) Когда весь бензол прибавлен, колбу помещают на водяную баню, нагретую до 60°, и ведут реакцию при этой температуре в течение получаса, часто и энергично перемешивая жидкость.
Затем реакционную смесь переливают в литровую колбу, содержащую 300 мл воды, перемешивают жидкость, охлаждают и при помощи делительной воронки отделяют находящийся в нижнем слое нитробензол*1. Его промывают в делительной воронке сначала разбавленным раствором углекислого натрия, а затем чистой водой.
Промытый нитробензол переливают в небольшую колбу и прибавляют прокаленный хлористый кальций. Колбу закрывают пробкой, в которую вставлена стеклянная трубка (в качестве обратного воздушного холодильника), и нагревают на водяной бане. Когда жидкость станет прозрачной, переливают нитробензол в перегонную колбу и перегоняют с воздушным холодильником. После отгонки небольшого количества непрореагировавшего бензола перегоняется нитробензол при температуре 204-207°. Отгонять продукт досуха не следует во избежание разложения остающегося в колбе динитробензола*2.
Выход около 22 г.
Темп. кип. чистого нитробензола 210,9°; уд. вес
1,2055; показатель преломления 1,5532.Препарат может быть использован для получения анилина или динитробензола.
Согласно правилу замещения в бензольном ядре, вторая нитрогруппа вступает в мета-положение по отношению к первой:
 Так как замещение второго водородного атома бензольного ядра на нитрогруппу идет значительно труднее, чем замещение первого, то при получении динитробензола приходится создавать более жесткие условия нитрования (повышенная концентрация кислот, более высокая температура).
При реакции, кроме основного продукта м-динитробензола, получаются небольшие количества п-динитробензола (около 3%) и о-динитробензола (около 1%), которые можно удалить путем перекристаллизации препарата из спирта.
В колбу емкостью 200 мл вливают нитробензол и нагревают под тягой на кипящей водяной бане. К горячему нитробензолу при энергичном перемешивании небольшими порциями прибавляют нитрующую смесь. Температура реакционной смеси во время нитрования не должна подниматься выше 115°.
После внесения всего количества нитрующей смеси нагревание и перемешивание продолжают еще в течение 30-40 мин. Конец реакции устанавливают на основании следующей пробы: каплю раствора вносят в пробирку с водой; динитробензол должен при этом выпадать в виде бледножелтых кристаллов; если этого не происходит, нагревание необходимо продолжить.
По окончании реакции смесь охлаждают до 70° и при энергичном перемешивании (под тягой) выливают в 100 мл холодной воды. Сырой динитробензол выпадает в виде аморфной массы. По охлаждении кислый раствор декантацией сливают с осадка*1, добавляют к последнему 50 мл воды и нагревают до кипения; динитробензол при этом плавится. По охлаждении воду сливают и повторяют ту же операцию, добавляя к воде углекислый натрий до резко щелочной реакции (по лакмусу). Охладив раствор, сливают воду через фильтр, а оставшийся на дне стакана динитробензол (в виде твердой лепешки) еще два раза плавят в чистой воде, беря каждый раз по 50 мл воды и сливая охлажденный раствор через тот же фильтр. Небольшое количество задержанных фильтром кристаллов промывают холодной водой, отжимают между листами фильтровальной бумаги, присоединяют к основной массе динитробензола, который вынимают из стакана, и высушивают на воздухе.
Выход около 15 г.
Полученный продукт плавится около 80°. Для получения вполне чистого м-динитробензола его перекристаллизовывают из спирта. Он образует бесцветные длинные иглы с темп. пл. 90°.
м-Динитробензол может быть использован для получения м-нитроанилина.
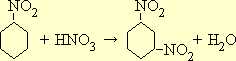
При нитровании нафталина нитрогруппа вступает всегда в a-положение:
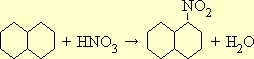 Нафталин нитруется значительно легче по сравнению с бензолом, и при несоблюдении надлежащих условий реакции наряду с мононитропродуктом могут получиться в значительном количестве 1,5- и 1,8-динитронафталин. Так как нафталин нитруется при температуре, лежащей ниже температуры его плавления, перед реакцией следует тщательно измельчить его; в противном случае выход продукта окажется пониженным.
Серную кислоту смешивают с 7 мл воды и с указанным количеством азотной кислоты. К смеси, нагретой до 50°, прибавляют тонко растертый нафталин и, поддерживая указанную температуру, ведут реакцию в течение 1 часа при постоянном перемешивании. (Целесообразно применить механическую мешалку.) Затем повышают температуру до 60° и в течение часа продолжают перемешивание смеси.
По охлаждении нитронафталин застывает в виде лепешки, плавающей на поверхности раствора. Кислую жидкость сливают, а сырой нитронафталин плавят в кипящей воде. По охлаждении воду сливают и повторяют эту операцию еще два раза. При такой обработке большая часть непрореагировавшего нафталина улетучивается с парами воды. Расплавленный продукт при энергичном перемешивании выливают в холодную воду, в которой он застывает в виде маленьких шариков. Осадок отфильтровывают, отжимают между листами фильтровальной бумаги и сушат на воздухе.
Выход около 15 г.
Полученный продукт не вполне чист и содержит небольшие количества динитронафталина и непрореагировавшего нафталина. Для получения чистого препарата его кристаллизуют из метилового спирта: он выпадает в виде желтых игл с темп. пл. 61,5°.
Для получения нафтиламина можно применять полученный препарат непосредственно, без кристаллизации.
Фенол нитруется очень легко уже на холоду под действием разбавленной азотной кислоты. В соответствии с ориентирующим влиянием OH-группы как заместителя 1-го рода при этом получается о- и п-нитрофенол:
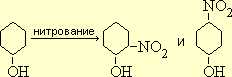 Разделение этих изомеров основано на том, что о-нитрофенол, в отличии от п-нитрофенола, перегоняется с водяным паром.
При нитровании фенола нужно избегать повышения температуры реакционной смеси во избежание образования ди- и тринитрофенола.
К фенолу прибавляют 5 мл воды, нагревают до плавления, и смесь постепенно при перемешивании вносят в азотную кислоту. Колбу с азотной кислотой охлаждают водой, наблюдая за тем, чтобы температура реакционной смеси все время была ниже 20°. Смесь, принимающую темную окраску, оставляют стоять в течение нескольких часов в холодной воде, периодически взбалтывая.
По окончании реакции тщательно сливают кислоту, промывают несколько раз водой оставшуюся в колбе маслянистую, частично осмолившуюся массу и подвергают ее перегонке с водяным паром (прибор собирают, как показано на рис. 18). В приемник в виде желтого быстро кристаллизующегося масла переходит о-нитрофенол. Если о-нитрофенол начинает кристаллизоваться в холодильнике, то на некоторое время прекращают подачу в него воды; горячий конденсат расплавляет кристаллы о-нитрофенола, и он переходит в приемник.
Выпавший в приемнике о-нитрофенол отфильтровывают на воронке Бюхнера, отжимают между листами фильтровальной бумаги и высушивают на воздухе.
Выход около 9 г.
Темп. пл. чистого о-нитрофенола 45°.
Если полученный продукт плавится при более низкой температуре, его перекристаллизовывают из метилового спирта.
Для выделения п-нитрофенола оставшуюся в колбе смолистую массу кипятят с 170 мл 10%-ного раствора едкого натра и небольшим количеством активного угля и фильтруют.
Еще горячий темный фильтрат упаривают до тех пор, пока капля раствора по охлаждении не будет застывать. Раствор охлаждают, выделившийся п-нитрофенолят отсасывают, промывают несколько раз небольшими порциями 10%-ного раствора едкого натра и хорошо отжимают на фильтре.
Полученную соль переносят в стакан и при нагревании разлагают 10%-ной соляной кислотой. Выделившийся нитрофенол по охлаждении застывает. Водный слой сливают и перекристаллизовывают нитрофенол из горячей 1-2%-ной соляной кислоты.
При охлаждении раствора п-нитрофенол выделяется в виде длинных бесцветных игл.
Выход 3,5-4 г.
Темп. пл. 114°.
Свободный анилин при нитровании легко подвергается окислению и осмолению. Для предохранения аминогруппы от окисления анилин сначала подвергают ацетилированию. Полученный ацетанилид нитруется с образованием преимущественно п-нитроацетанилида:
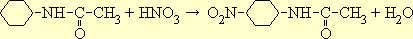 Если нитрование ведется при низкой температуре, то изомерный о-нитроацетанилид образуется лишь в весьма небольших количествах; повышенная же температура благоприятствует его образованию.
При нагревании со щелочью п-нитроацетанилид подвергается омылению и дает п-нитроанилин:
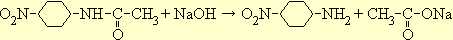
Тонко измельченный сухой ацетанилид вносят в 30 мл концентрированной серной кислоты и перемешивают до тех пор, пока не получится вполне прозрачный раствор. Температура при этом не должна подниматься выше 25° во избежание омыления ацетанилида.
Раствор охлаждают до 0° в смеси льда и соли, и постепенно приливают смесь 8 мл азотной кислоты и 5 мл концентрированной серной кислоты. Температура во время нитрования не должна превышать 2-3° во избежание образования значительных количеств о-нитросоединения. После того как прибавлена вся кислота, продолжают перемешивание еще в течение получаса и смесь оставляют стоять на холоду в течение ночи.
На следующий день вливают раствор в смесь 35 мл воды и 35 г толченого льда; тотчас выпадает нитроацетанилид. Через полчаса осадок отфильтровывают, хорошо промывают водой, переносят в стакан с 50 мл воды, прибавляют углекислый натрий до щелочной реакции (по лакмусу) и нагревают до кипения. При этой обработке о-нитроацетанилид омыляется, а п-нитроацетанилид остается без изменения. Охлаждают раствор до 50°, отфильтровывают кристаллы п-нитроацетанилида, хорошо промывают водой и высушивают на воздухе.
Выход около 16 г.
Темп. пл. 207°.
Если полученный препарат недостаточно чист, его можно перекристаллизовать из спирта.
Для получения п-нитроанилина сырой п-нитроацетанилид смешивают с 20 мл воды, приливают 12 мл 35%-ного раствора едкого натра и кипятят до тех пор, пока капля раствора, внесенная в 10%-ную соляную кислоту, не будет растворяться без осадка.
Обычно омыление заканчивается через 2,5-3 часа. Нужно следить, чтобы во время кипячения реакция все время оставалась щелочной. Смеси дают охладиться до 40°, отфильтровывают осадок, тщательно промывают холодной водой и высушивают.
Выход 12-13 г.
Темп. пл. 148°.
Препарат может быть использован для получения п-нитрофенилгидразина.
*1Остающийся в делительной воронке водный слой, содержащий серную и азотную кислоты (а также аналогичные ему растворы), нельзя непосредственно выливать в канализационную раковину; предварительно его необходимо нейтрализовать известковым молоком или известью.
*2Лучше перегонять в вакууме.
| |